matchms documentation
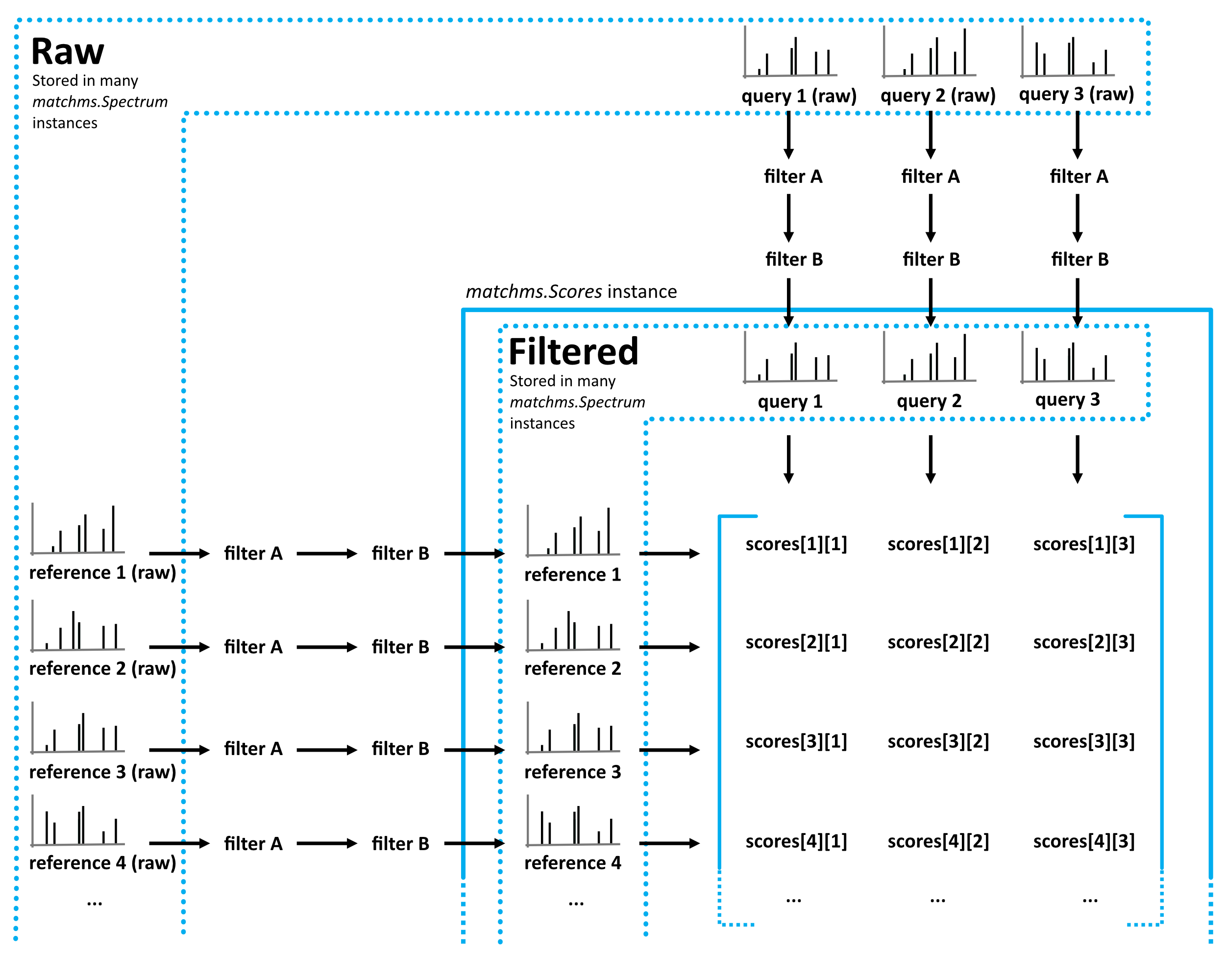
Matchms is an open-access Python library to import, process, clean, and compare tandem mass spectrometry (MS/MS) data. It helps you build reproducible workflows from raw spectra to similarity scores and downstream analysis.
Get started
Install via conda (recommended):
conda create -n matchms python=3.12
conda activate matchms
conda install -c conda-forge -c bioconda matchms