matchms package
- class matchms.Fingerprints(fingerprint_algorithm: str = 'daylight', fingerprint_method: str = 'bit', nbits: int = 2048, ignore_stereochemistry: bool = False, **kwargs)[source]
Bases:
objectComputes and stores inchikey-fingerprint mapping for a list of spectra,
For example
from matchms import Fingerprints from matchms import Spectrum import numpy as np spectrum_1 = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.7, 0.2, 0.1]), metadata={"inchikey": "OTMSDBZUPAUEDD-UHFFFAOYSA-N", "smiles":"CC", "precursor_mz": 150.0}) spectrum_2 = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.7, 0.2, 0.1]), metadata={"inchikey": "UGFAIRIUMAVXCW-UHFFFAOYSA-N", "smiles": "[C-]#[O+]", "precursor_mz": 150.0}) spectra = [spectrum_1, spectrum_2] fpgen = Fingerprints() fpgen.compute_fingerprints(spectra) print(fpgen.fingerprint_count) print(type(fpgen.get_fingerprint_by_inchikey('OTMSDBZUPAUEDD-UHFFFAOYSA-N')))
Should output
2 <class 'numpy.ndarray'>
- config
The configuration for the fingerprints e.g., used algorithm, nbits, …
- fingerprints
The computed fingerprints. Use after compute_fingerprints().
- fingerprints_count
The number of fingerprints computed.
- to_dataframe
A DataFrame containing the inchikey and fingerprint
- __init__(fingerprint_algorithm: str = 'daylight', fingerprint_method: str = 'bit', nbits: int = 2048, ignore_stereochemistry: bool = False, **kwargs)[source]
- Parameters:
fingerprint_algorithm – The fingerprint algorithm to use. Available options: daylight, morgan1, morgan2, morgan3.
fingerprint_method – The fingerprint method to use. Available options: bit, sparse_bit, count, sparse_count.
nbits – The number of bits or fingerprint size. Defaults to 2048.
ignore_stereochemistry – Determines which inchikey version will be used. If set to true the first 14 chars of the inchikey are used.
- compute_fingerprint(spectrum: Spectrum) ndarray | None[source]
Computes a single fingerprint for a given spectrum.
- Parameters:
spectrum – A spectrum for which a fingerprint is to be calculated.
Return
--------------
Optional[np.ndarray] – The corresponding fingerprint.
- compute_fingerprints(spectra: list[Spectrum])[source]
Computes fingerprints for a list of spectra.
This will first create a dict with unique spectra and then computes fingerprints for all mols. Only valid fingerprints will be added to the mapping. Query specific fingerprints by using get_fingerprint_by_spectrum() or get_fingerprint_by_inchikey()
- Parameters:
spectra – List of Spectrum
- class matchms.Fragments(mz=None, intensities=None)[source]
Bases:
objectStores arrays of intensities and M/z values, with some checks on their internal consistency.
For example
import numpy as np from matchms import Fragments mz = np.array([10, 20, 30], dtype="float") intensities = np.array([100, 20, 300], dtype="float") peaks = Fragments(mz=mz, intensities=intensities) print(peaks[2])
Should output
[ 30. 300.]
- mz
Numpy array of m/z values.
- intensities
Numpy array of peak intensity values.
- property intensities
getter method for intensities private variable
- property mz
getter method for mz private variable
- property to_numpy
getter method to return stacked numpy array of both peak mz and intensities
- class matchms.Metadata(metadata: dict = None, matchms_key_style: bool = True)[source]
Bases:
objectClass to handle spectrum metadata in matchms.
Metadata entries will be stored as PickyDict dictionary in metadata.data. Unlike normal Python dictionaries, not all key names will be accepted. Key names will be forced to be lower-case to avoid confusions between key such as “Precursor_MZ” and “precursor_mz”.
To avoid the default harmonization of the metadata dictionary use the option matchms_key_style=False.
Code example:
metadata = Metadata({"Precursor_MZ": 201.5, "Compound Name": "SuperStuff"}) print(metadata["precursor_mz"]) # => 201.5 print(metadata["compound_name"]) # => SuperStuff
Or if the matchms default metadata harmonization should not take place:
metadata = Metadata({"Precursor_MZ": 201.5, "Compound Name": "SuperStuff"}, matchms_key_style=False) print(metadata["precursor_mz"]) # => 201.5 print(metadata["compound_name"]) # => None (now you need to use "compound name")
- __init__(metadata: dict = None, matchms_key_style: bool = True)[source]
- Parameters:
metadata – Spectrum metadata as a dictionary.
matchms_key_style – Set to False if metadata harmonization to default keys is not desired. The default is True.
- harmonize_keys()[source]
Runs default harmonization of metadata.
Method harmonized metadata field names which include setting them to lower-case and running a series of regex replacements followed by the default field name replacements (such as precursor_mass –> precursor_mz).
- harmonize_values()[source]
Runs default harmonization of metadata.
This includes harmonizing entries for ionmode, retention time and index, charge, as well as the removal of invalid entries (“”, “NA”, “N/A”, “NaN”).
- static set_key_replacements(keys: dict)[source]
Set key replacements for metadata harmonization.
- Parameters:
keys – Dictionary with key replacements.
- class matchms.Pipeline(workflow: OrderedDict, progress_bar=True, logging_level: str = 'WARNING', logging_file: str | None = None)[source]
Bases:
objectCentral pipeline class.
The matchms Pipeline class is meant to make running extensive analysis pipelines fast and easy. It can be used in two different ways. First, a pipeline can be defined using a config file (a yaml file, best to start from the template provided to define your own pipline).
Once a config file is defined, the pipeline can be executed with the following code:
The second way to define a pipeline is via a Python script. The following code is an example of how this works:
To combine this with custom made scores or available matchms-compatible scores such as Spec2Vec or MS2DeepScore, it is also possible to pass objects instead of names to create_workflow
from spec2vec import Spec2Vec workflow = create_workflow( score_computations=[ ["precursormzmatch", {"tolerance": 120.0}], [Spec2Vec, {"model": "my_spec2vec_model.model"}], ["filter_by_range", {"name": "Spec2Vec", "low": 0.3}], ] )
- __init__(workflow: OrderedDict, progress_bar=True, logging_level: str = 'WARNING', logging_file: str | None = None)[source]
- Parameters:
workflow – Contains an orderedDict containing the workflow settings. Can be created using create_workflow.
progress_bar – Default is True. Set to False if no progress bar should be displayed.
- import_spectra(query_files: List[str] | str, reference_files: List[str] | str | None = None)[source]
Import spectra from file(s).
- Parameters:
query_files – List of files, or single filename, containing the query spectra.
reference_files – List of files, or single filename, containing the reference spectra. If set to None (default) then all query spectra will be compared to each other.
- import_spectrums(query_files: List[str] | str, reference_files: List[str] | str | None = None)[source]
Wrapper method for import_spectra()
- Parameters:
query_files – List of files, or single filename, containing the query spectra.
reference_files – List of files, or single filename, containing the reference spectra. If set to None (default) then all query spectra will be compared to each other.
- run(query_files, reference_files=None, cleaned_query_file=None, cleaned_reference_file=None, create_report=True)[source]
Execute the defined Pipeline workflow.
This method will execute all steps of the workflow. 1) Initializing the log file and importing the spectra 2) Spectrum processing (using matchms filters) 3) Score Computations
- class matchms.Scores(references: List[object] | Tuple[object] | ndarray, queries: List[object] | Tuple[object] | ndarray, is_symmetric: bool = False)[source]
Bases:
objectContains reference and query spectra and the scores between them.
The scores can be retrieved as a matrix with the
Scores.scoresattribute. The reference spectrum, query spectrum, score pairs can also be iterated over in query then reference order.Example to calculate scores between 2 spectra and iterate over the scores
import numpy as np from matchms import calculate_scores from matchms import Spectrum from matchms.similarity import CosineGreedy spectrum_1 = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.7, 0.2, 0.1]), metadata={'id': 'spectrum1'}) spectrum_2 = Spectrum(mz=np.array([100, 140, 190.]), intensities=np.array([0.4, 0.2, 0.1]), metadata={'id': 'spectrum2'}) spectrum_3 = Spectrum(mz=np.array([110, 140, 195.]), intensities=np.array([0.6, 0.2, 0.1]), metadata={'id': 'spectrum3'}) spectrum_4 = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.6, 0.1, 0.6]), metadata={'id': 'spectrum4'}) references = [spectrum_1, spectrum_2] queries = [spectrum_3, spectrum_4] similarity_measure = CosineGreedy() scores = calculate_scores(references, queries, similarity_measure) for (reference, query, score) in scores: print(f"Cosine score between {reference.get('id')} and {query.get('id')}" + f" is {score[0]:.2f} with {score[1]} matched peaks")
Should output
Cosine score between spectrum1 and spectrum4 is 0.80 with 3 matched peaks Cosine score between spectrum2 and spectrum3 is 0.14 with 1 matched peaks Cosine score between spectrum2 and spectrum4 is 0.61 with 1 matched peaks
- __init__(references: List[object] | Tuple[object] | ndarray, queries: List[object] | Tuple[object] | ndarray, is_symmetric: bool = False)[source]
- Parameters:
references – List of reference objects
queries – List of query objects
is_symmetric – Set to True when references and queries are identical (as for instance for an all-vs-all comparison). By using the fact that score[i,j] = score[j,i] the calculation will be about 2x faster. Default is False.
- calculate(similarity_function: BaseSimilarity, name: str = None, array_type: str = 'numpy', join_type='left') Scores[source]
Calculate the similarity between all reference objects vs all query objects using the most suitable available implementation of the given similarity_function. If Scores object already contains similarity scores, the newly computed measures will be added to a new layer (name –> layer name). Additional scores will be added as specified with join_type, the default being ‘left’.
- Parameters:
similarity_function – Function which accepts a reference + query object and returns a score or tuple of scores
name – Label of the new scores layer. If None, the name of the similarity_function class will be used.
array_type – Specify the type of array to store and compute the scores. Choose from “numpy” or “sparse”.
join_type – Choose from left, right, outer, inner to specify the merge type.
- filter_by_range(**kwargs)[source]
Remove all scores for which the score name is outside the given range.
- Parameters:
kwargs – See “Keyword arguments” section below.
- Keyword Arguments:
name – Name of the score which is used for filtering. Run .score_names to see all scores stored in the sparse array.
low – Lower threshold below which all scores will be removed.
high – Upper threshold above of which all scores will be removed.
above_operator – Define operator to be used to compare against low. Default is ‘>’. Possible choices are ‘>’, ‘<’, ‘>=’, ‘<=’.
below_operator – Define operator to be used to compare against high. Default is ‘<’. Possible choices are ‘>’, ‘<’, ‘>=’, ‘<=’.
- scores_by_query(query: List[object] | Tuple[object] | ndarray, name: str = None, sort: bool = False) ndarray[source]
Return all scores for the given query spectrum.
For example
import numpy as np from matchms import calculate_scores, Scores, Spectrum from matchms.similarity import CosineGreedy spectrum_1 = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.7, 0.2, 0.1]), metadata={'id': 'spectrum1'}) spectrum_2 = Spectrum(mz=np.array([100, 140, 190.]), intensities=np.array([0.4, 0.2, 0.1]), metadata={'id': 'spectrum2'}) spectrum_3 = Spectrum(mz=np.array([110, 140, 195.]), intensities=np.array([0.6, 0.2, 0.1]), metadata={'id': 'spectrum3'}) spectrum_4 = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.6, 0.1, 0.6]), metadata={'id': 'spectrum4'}) references = [spectrum_1, spectrum_2, spectrum_3] queries = [spectrum_2, spectrum_3, spectrum_4] scores = calculate_scores(references, queries, CosineGreedy()) selected_scores = scores.scores_by_query(spectrum_4, 'CosineGreedy_score', sort=True) print([float(x[1][0].round(3)) for x in selected_scores])
Should output
[0.796, 0.613]
- Parameters:
query – Single query Spectrum.
name – Name of the score that should be returned (if multiple scores are stored).
sort – Set to True to obtain the scores in a sorted way (relying on the
sort()function from the given similarity_function).
- scores_by_reference(reference: List[object] | Tuple[object] | ndarray, name: str = None, sort: bool = False) ndarray[source]
Return all scores of given name for the given reference spectrum.
- Parameters:
reference – Single reference Spectrum.
name – Name of the score that should be returned (if multiple scores are stored).
sort – Set to True to obtain the scores in a sorted way (relying on the
sort()function from the given similarity_function).
- to_array(name=None) ndarray[source]
Scores as numpy array
For example
import numpy as np from matchms import calculate_scores, Scores, Spectrum from matchms.similarity import IntersectMz spectrum_1 = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.7, 0.2, 0.1])) spectrum_2 = Spectrum(mz=np.array([100, 140, 190.]), intensities=np.array([0.4, 0.2, 0.1])) spectra = [spectrum_1, spectrum_2] scores = calculate_scores(spectra, spectra, IntersectMz()).to_array() print(scores.shape) print(scores.tolist())
Should output
(2, 2) [[1.0, 0.2], [0.2, 1.0]]
- Parameters:
name – Name of the score that should be returned (if multiple scores are stored).
- to_coo(name=None) coo_matrix[source]
Scores as scipy sparse COO matrix
- Parameters:
name – Name of the score that should be returned (if multiple scores are stored).
- class matchms.Spectrum(mz: array, intensities: array, metadata: dict | None = None, metadata_harmonization: bool = True)[source]
Bases:
objectContainer for a collection of peaks, losses and metadata.
Spectrum peaks are stored as
Fragmentsobject which can be addressed calling spectrum.peaks and contains m/z values and the respective peak intensities.Spectrum metadata is stored as
Metadataobject which can be addressed by spectrum.metadata.Code example
import numpy as np from matchms import Scores, Spectrum from matchms.similarity import CosineGreedy spectrum = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.7, 0.2, 0.1]), metadata={"id": 'spectrum1', "precursor_mz": 222.333, "peak_comments": {200.: "the peak at 200 m/z"}}) print(spectrum) print(spectrum.peaks.mz[0]) print(spectrum.peaks.intensities[0]) print(spectrum.get('id')) print(spectrum.peak_comments.get(200))
Should output
Spectrum(precursor m/z=222.33, 3 fragments between 100.0 and 200.0) 100.0 0.7 spectrum1 the peak at 200 m/z
- losses
Losses of spectrum, the difference between the precursor and all peaks.
Can be filled with
from matchms import Fragments spectrum.losess = Fragments(mz=np.array([50.]), intensities=np.array([0.1]))
- Type:
Fragments or None
- __init__(mz: array, intensities: array, metadata: dict | None = None, metadata_harmonization: bool = True)[source]
- Parameters:
mz – Array of m/z for the peaks
intensities – Array of intensities for the peaks
metadata – Dictionary with for example the scan number of precursor m/z.
metadata_harmonization (bool, optional) – Set to False if default metadata filters should not be applied. The default is True.
- compute_losses(loss_mz_from: float = 0.0, loss_mz_to: float = None) Fragments | None[source]
This will compute the “losses”, i.e. the differences between the precursor_mz and the individual fragment m/z values. Only losses between loss_mz_from and loss_mz_to will be kept.
- Parameters:
loss_mz_from – Float value to set the minimum acceptable loss value. Default is 0.0.
loss_mz_to – Float value to set the maximum acceptable loss value. Default is None which means that the los_mz_to will be set to the spectrum’s precursor_mz.
- get(key: str, default=None)[source]
Retrieve value from
metadatadict. Shorthand forval = self.metadata[key]
- metadata_dict(export_style: str = 'matchms') dict[source]
Convert spectrum metadata to Python dictionary.
- Parameters:
export_style – Converts the keys to the required export style. One of [“matchms”, “massbank”, “nist”, “riken”, “gnps”]. Default is “matchms”
- metadata_hash()[source]
Return a (truncated) sha256-based hash which is generated based on the spectrum metadata. Spectra with same metadata results in same metadata_hash.
- plot(figsize=(8, 6), dpi=200, **kwargs)[source]
Plot to visually inspect a spectrum run
spectrum.plot()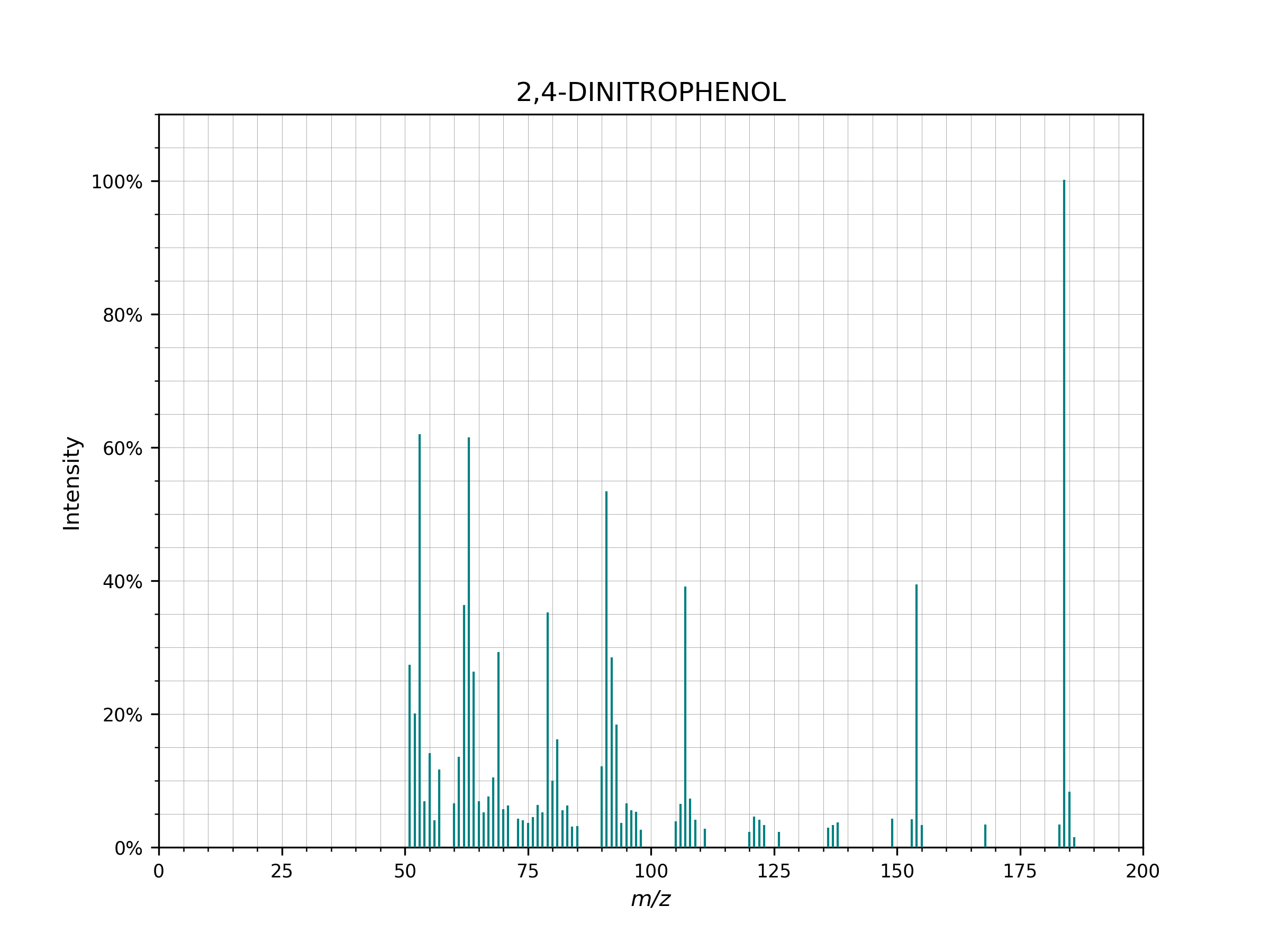
Example of a spectrum plotted using
spectrum.plot()..
- plot_against(other_spectrum, figsize=(8, 6), dpi=200, **spectrum_kws)[source]
Compare two spectra visually in a mirror plot.
To visually compare the peaks of two spectra run
spectrum.plot_against(other_spectrum)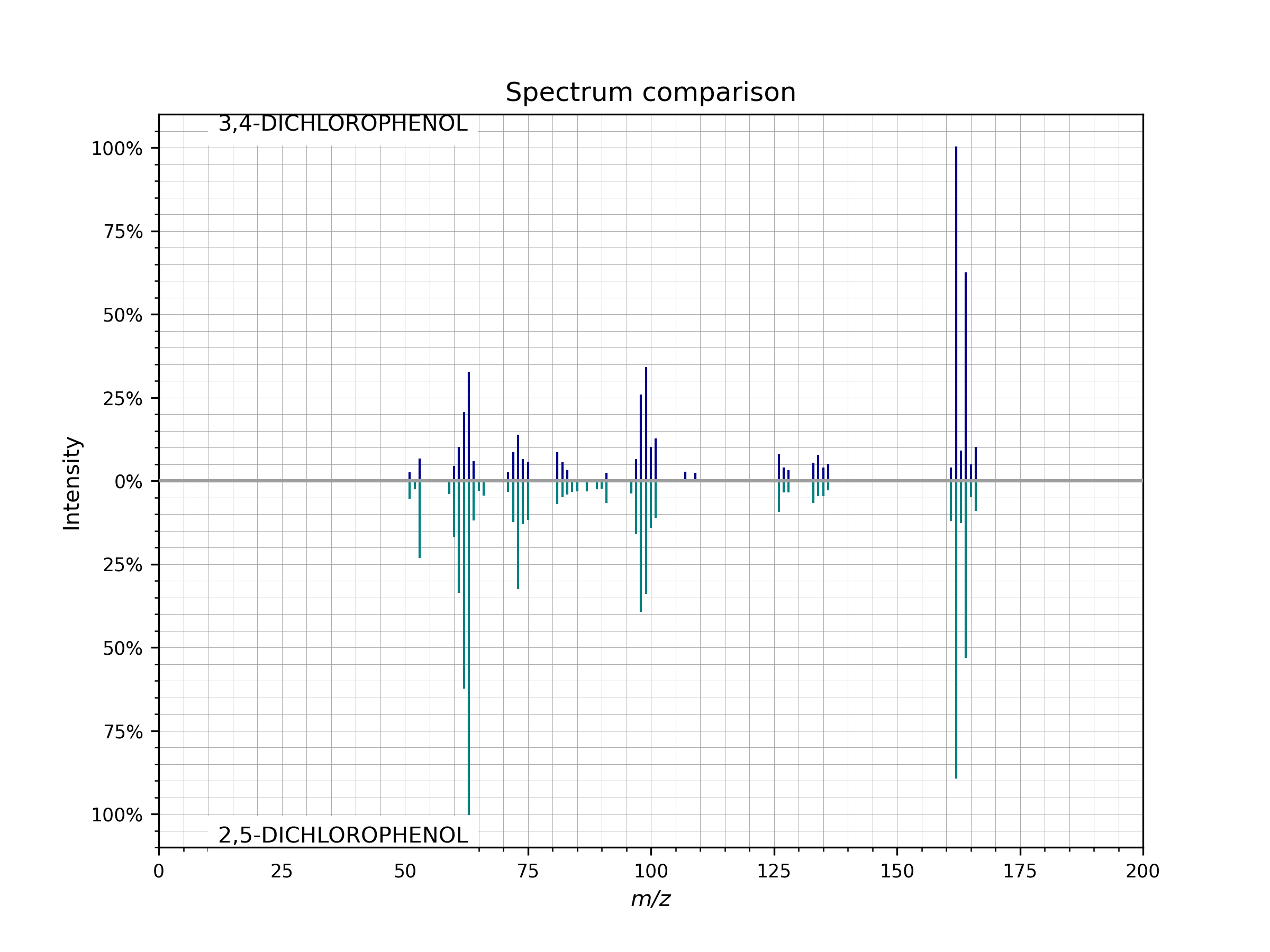
Example of a mirror plot comparing two spectra
spectrum.plot_against()..
- spectrum_hash()[source]
Return a (truncated) sha256-based hash which is generated based on the spectrum peaks (mz:intensity pairs). Spectra with same peaks will results in same spectrum_hash.
- class matchms.SpectrumProcessor(filters: Iterable[str | Callable | Tuple[Callable | str, Dict[str, any]]])[source]
Bases:
objectA class to process spectra using a series of filters.
The class enables a user to define a custom spectrum processing workflow by setting multiple flags and parameters.
- Parameters:
filters – A list of filter functions, see add_filter for all the allowed formats.
- parse_and_add_filter(filter_description: str | Callable | Tuple[Callable | str, Dict[str, any]], filter_position: int | None = None)[source]
Adds a filter, by parsing the different allowed inputs.
- filter:
Allowed formats: str (has to be a matchms function name) (str, {str, any} (has to be a matchms function name, followed by parameters) Callable (can be matchms filter or custom made filter) Callable, {str, any} (the dict should be parameters.
- filter_position:
If None: Matchms filters are automatically ordered. Custom filters will be added at the end of the filter list. If not None, the filter will be added to the given position in the filter order list.
- process_spectra(spectra: list, progress_bar: bool = True, cleaned_spectra_file=None, create_report: bool | None = False)[source]
Process a list of spectra with all filters in the processing pipeline.
- Parameters:
create_report (bool, optional) – Creates and outputs a report of the main changes during processing. The report will be returned as pandas DataFrame. Default is set to False.
progress_bar (bool, optional) – Displays progress bar if set to True. Default is True.
cleaned_spectra_file – Path to where the cleaned spectra should be saved.
- Returns:
spectra – List containing the processed spectra.
processing_report – A ProcessingReport containing the effect of the filters.
- process_spectrum(spectrum, processing_report: ProcessingReport | None = None)[source]
Process the given spectrum with all filters in the processing pipeline.
- process_spectrums(spectra: list, progress_bar: bool = True, cleaned_spectra_file=None, create_report: bool | None = False)[source]
Wrapper method for process_spectra()
- Parameters:
create_report (bool, optional) – Creates and outputs a report of the main changes during processing. The report will be returned as pandas DataFrame. Default is set to False.
progress_bar (bool, optional) – Displays progress bar if set to True. Default is True.
cleaned_spectra_file – Path to where the cleaned spectra should be saved.
- Returns:
spectra – List containing the processed spectra.
processing_report – A ProcessingReport containing the effect of the filters.
- matchms.calculate_scores(references: List[object] | Tuple[object] | ndarray, queries: List[object] | Tuple[object] | ndarray, similarity_function: BaseSimilarity, array_type: str = 'numpy', is_symmetric: bool = False) Scores[source]
Calculate the similarity between all reference objects versus all query objects.
Example to calculate scores between 2 spectra and iterate over the scores
import numpy as np from matchms import calculate_scores, Spectrum from matchms.similarity import CosineGreedy spectrum_1 = Spectrum(mz=np.array([100, 150, 200.]), intensities=np.array([0.7, 0.2, 0.1]), metadata={'id': 'spectrum1'}) spectrum_2 = Spectrum(mz=np.array([100, 140, 190.]), intensities=np.array([0.4, 0.2, 0.1]), metadata={'id': 'spectrum2'}) spectra = [spectrum_1, spectrum_2] scores = calculate_scores(spectra, spectra, CosineGreedy()) for (reference, query, score) in scores: print(f"Cosine score between {reference.get('id')} and {query.get('id')}" + f" is {score[0]:.2f} with {score[1]} matched peaks")
Should output
Cosine score between spectrum1 and spectrum1 is 1.00 with 3 matched peaks Cosine score between spectrum1 and spectrum2 is 0.83 with 1 matched peaks Cosine score between spectrum2 and spectrum1 is 0.83 with 1 matched peaks Cosine score between spectrum2 and spectrum2 is 1.00 with 3 matched peaks
- Parameters:
references – List of reference objects
queries – List of query objects
similarity_function – Function which accepts a reference + query object and returns a score or tuple of scores
array_type – Specify the type of array to store and compute the scores. Choose from “numpy” or “sparse”.
is_symmetric – Set to True when references and queries are identical (as for instance for an all-vs-all comparison). By using the fact that score[i,j] = score[j,i] the calculation will be about 2x faster. Default is False.
- Return type:
- matchms.set_matchms_logger_level(loglevel: str, logger_name='matchms')[source]
Update logging level to given loglevel.
- Parameters:
loglevels – Can be ‘DEBUG’, ‘INFO’, ‘WARNING’, ‘ERROR’, ‘CRITICAL’.
logger_name – Default is “matchms”. Change if logger name should be different.
Subpackages
- matchms.exporting package
- Functions for exporting mass spectral data
save_as_mzspeclib()- Submodules
- matchms.filtering package
- Processing (or: filtering) mass spectra
SpeciesStringSpeciesString.dirtySpeciesString.targetSpeciesString.cleanedSpeciesString.__init__()SpeciesString.clean()SpeciesString.clean_as_inchi()SpeciesString.clean_as_inchikey()SpeciesString.clean_as_smiles()SpeciesString.guess_target()SpeciesString.looks_like_a_smiles()SpeciesString.looks_like_an_inchi()SpeciesString.looks_like_an_inchikey()
add_compound_name()add_fingerprint()add_parent_mass()add_precursor_formula()add_precursor_mz()add_retention_index()add_retention_time()clean_adduct()clean_compound_name()correct_charge()default_filters()derive_adduct_from_name()derive_annotation_from_compound_name()derive_formula_from_name()derive_formula_from_smiles()derive_inchi_from_smiles()derive_inchikey_from_inchi()derive_ionmode()derive_smiles_from_inchi()harmonize_undefined_inchi()harmonize_undefined_inchikey()harmonize_undefined_smiles()interpret_pepmass()make_charge_int()normalize_intensities()reduce_to_number_of_peaks()remove_noise_below_frequent_intensities()remove_peaks_around_precursor_mz()remove_peaks_outside_top_k()remove_peaks_relative_to_precursor_mz()remove_profiled_spectra()repair_adduct_and_parent_mass_based_on_smiles()repair_adduct_based_on_parent_mass()repair_inchi_inchikey_smiles()repair_not_matching_annotation()repair_parent_mass_from_smiles()repair_parent_mass_is_molar_mass()repair_parent_mass_match_smiles_wrapper()repair_smiles_of_salts()require_compound_name()require_correct_ionmode()require_formula()require_matching_adduct_precursor_mz_parent_mass()require_maximum_number_of_peaks()require_minimum_number_of_high_peaks()require_minimum_number_of_peaks()require_parent_mass_match_smiles()require_precursor_below_mz()require_precursor_mz()require_retention_index()require_retention_time()require_valid_annotation()select_by_intensity()select_by_mz()select_by_relative_intensity()- Subpackages
- Submodules
- matchms.importing package
- Functions for importing mass spectral data
load_from_json()load_from_mgf()load_from_msp()load_from_mzml()load_from_mzxml()load_from_pickle()load_from_usi()load_spectra()scores_from_json()scores_from_pickle()- Submodules
- matchms.importing.load_from_json module
- matchms.importing.load_from_mgf module
- matchms.importing.load_from_msp module
- matchms.importing.load_from_mzml module
- matchms.importing.load_from_mzxml module
- matchms.importing.load_from_pickle module
- matchms.importing.load_from_usi module
- matchms.importing.load_scores module
- matchms.importing.load_spectra module
- matchms.importing.parsing_utils module
- matchms.networking package
- matchms.plotting package
- matchms.reference_spectra package
aspirin()cocaine()glucose()hydroxy_cholesterol()phenylalanine()salicin()- Submodules
- matchms.similarity package
- Functions for computing spectra similarities
BinnedEmbeddingSimilarityBinnedEmbeddingSimilarity.__init__()BinnedEmbeddingSimilarity.build_ann_index()BinnedEmbeddingSimilarity.compute_embeddings()BinnedEmbeddingSimilarity.get_anns()BinnedEmbeddingSimilarity.get_embeddings()BinnedEmbeddingSimilarity.get_index_anns()BinnedEmbeddingSimilarity.keep_score()BinnedEmbeddingSimilarity.load_ann_index()BinnedEmbeddingSimilarity.load_embeddings()BinnedEmbeddingSimilarity.matrix()BinnedEmbeddingSimilarity.pair()BinnedEmbeddingSimilarity.save_ann_index()BinnedEmbeddingSimilarity.score_datatypeBinnedEmbeddingSimilarity.sparse_array()BinnedEmbeddingSimilarity.store_embeddings()BinnedEmbeddingSimilarity.to_dict()
BlinkCosineCosineGreedyCosineHungarianCosineLinearFingerprintSimilarityFlashSimilarityIntersectMzMetadataMatchModifiedCosineGreedyModifiedCosineHungarianNeutralLossesCosineParentMassMatchPrecursorMzMatch- Submodules
- matchms.similarity.BaseEmbeddingSimilarity module
- matchms.similarity.BaseSimilarity module
- matchms.similarity.BinnedEmbeddingSimilarity module
- matchms.similarity.BlinkCosine module
- matchms.similarity.CosineGreedy module
- matchms.similarity.CosineHungarian module
- matchms.similarity.CosineLinear module
- matchms.similarity.FingerprintSimilarity module
- matchms.similarity.FlashSimilarity module
- matchms.similarity.IntersectMz module
- matchms.similarity.MetadataMatch module
- matchms.similarity.ModifiedCosineGreedy module
- matchms.similarity.ModifiedCosineHungarian module
- matchms.similarity.NeutralLossesCosine module
- matchms.similarity.ParentMassMatch module
- matchms.similarity.PrecursorMzMatch module
- matchms.similarity.cosine_linear_functions module
- matchms.similarity.flash_utils module
- matchms.similarity.spectrum_similarity_functions module
- matchms.similarity.vector_similarity_functions module
Submodules
- matchms.Fingerprints module
- matchms.Fragments module
- matchms.Metadata module
- matchms.Pipeline module
- matchms.Scores module
- matchms.Spectrum module
SpectrumSpectrum.peaksSpectrum.lossesSpectrum.metadataSpectrum.__init__()Spectrum.clone()Spectrum.compute_losses()Spectrum.get()Spectrum.metadata_dict()Spectrum.metadata_hash()Spectrum.plot()Spectrum.plot_against()Spectrum.set()Spectrum.spectrum_hash()Spectrum.to_dict()Spectrum.update_peak_comments_mz_tolerance()
- matchms.calculate_scores module
- matchms.constants module
- matchms.hashing module
- matchms.logging_functions module
- matchms.typing module
- matchms.utils module
- matchms.yaml_file_functions module