matchms.filtering package
Processing (or: filtering) mass spectra
Provided functions will usually only perform a single action to a spectrum. This can be changes or corrections of metadata, or peak filtering. More complicated processing pipelines can be build by stacking several of the provided filters.
Because there are numerous filter functions in matchms and because they often need to be applied in a specific order, the most feasible workflow for users is to use the SpectrumProcessor class to define a spetrum processing pipeline. Here is an example:
import numpy as np
from matchms import Spectrum
from matchms import SpectrumProcessor
spectrum = Spectrum(mz=np.array([100, 120, 150, 200.]),
intensities=np.array([200.0, 300.0, 50.0, 1.0]),
metadata={'id': 'spectrum1'})
# Users can pick a predefined pipeline from default pipelines, or specify a list of filters
processing = SpectrumProcessor(["normalize_intensities"])
# Run the processing pipeline:
spectrum_filtered = processing.process_spectrum(spectrum)
max_intensity = spectrum_filtered.peaks.intensities.max()
print(f"Maximum intensity is {max_intensity:.2f}")
Should output
Maximum intensity is 1.00
It is also possible to run each filter function individually. This for instance makes sense if users want to develop a highly customized spectrum processing routine. Example of how to use a single filter function:
import numpy as np
from matchms import Spectrum
from matchms.filtering import normalize_intensities
spectrum = Spectrum(mz=np.array([100, 120, 150, 200.]),
intensities=np.array([200.0, 300.0, 50.0, 1.0]),
metadata={'id': 'spectrum1'})
spectrum_filtered = normalize_intensities(spectrum)
max_intensity = spectrum_filtered.peaks.intensities.max()
print(f"Maximum intensity is {max_intensity:.2f}")
Should output
Maximum intensity is 1.00
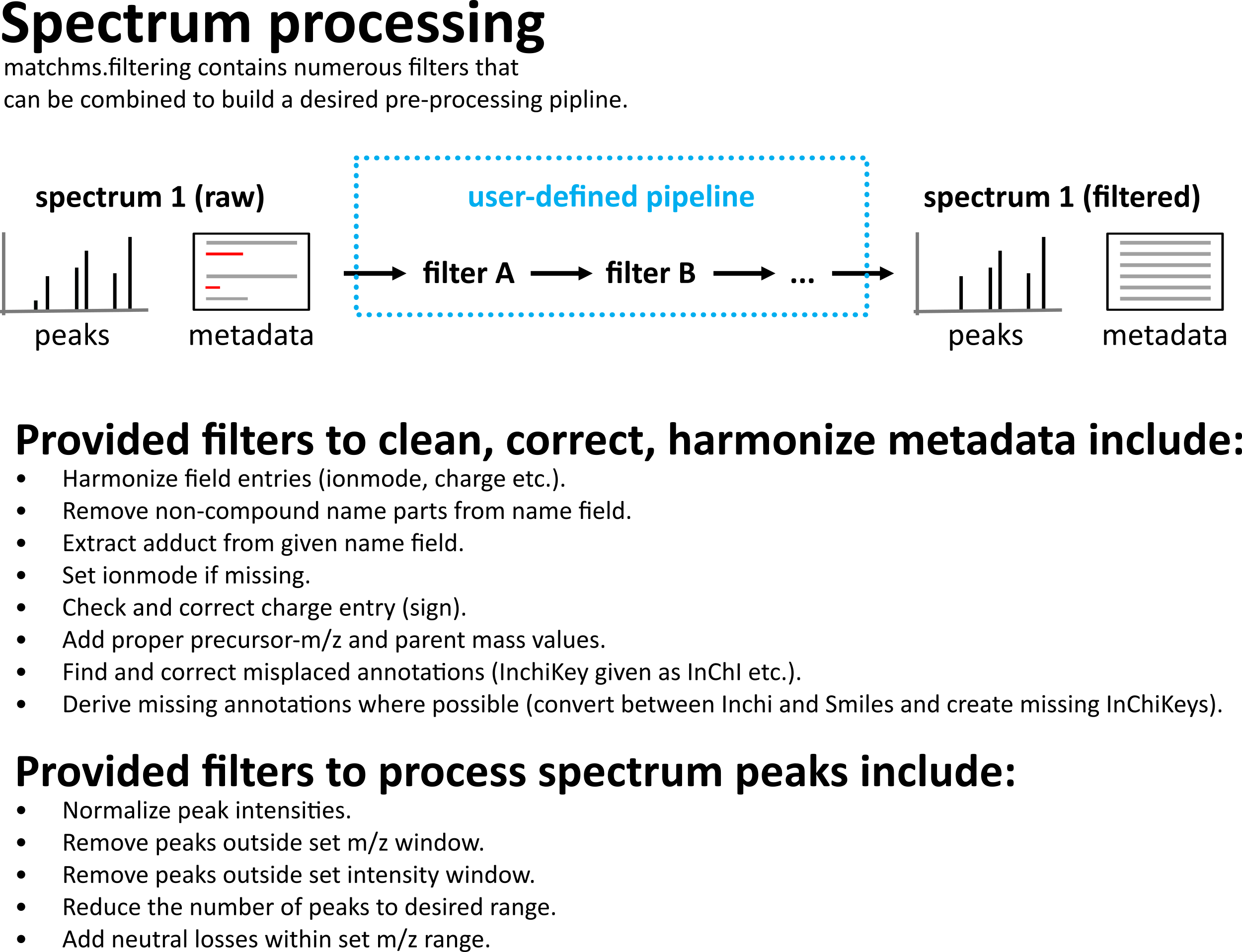
Sketch of matchms spectrum processing.
- class matchms.filtering.SpeciesString(dirty: str)[source]
Bases:
objectA class to process and clean different types of chemical structure strings including InChI, InChIKey, and SMILES.
The class takes a raw input string, determines the intended structure type, and then cleans the string based on its type.
- target
The intended structure type determined from the input string. Could be ‘inchi’, ‘inchikey’, ‘smiles’, or None if no valid type was identified.
- Type:
- matchms.filtering.add_compound_name(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Add compound_name to correct field: “compound_name” in metadata.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added compound name, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.add_fingerprint(spectrum_in: Spectrum | None, fingerprint_type: str = 'daylight', nbits: int = 2048, clone: bool | None = True) Spectrum | None[source]
Add molecular finterprint to spectrum.
If smiles or inchi present in metadata, derive a molecular finterprint and add it to the spectrum.
- Parameters:
spectrum_in – Input spectrum.
fingerprint_type – Determine method for deriving molecular fingerprints. Supported choices are “daylight”, “morgan1”, “morgan2”, “morgan3”. Default is “daylight”.
nbits – Dimension or number of bits of generated fingerprint. Default is 2048.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added fingerprint derived from SMILES or INCHI, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.add_parent_mass(spectrum_in: Spectrum, estimate_from_adduct: bool = True, overwrite_existing_entry: bool = False, estimate_from_charge: bool = True, clone: bool | None = True) Spectrum | None[source]
Add estimated parent mass to metadata (if not present yet).
Method to calculate the parent mass from given precursor m/z together with charge and/or adduct. Will take precursor m/z from “precursor_mz” as provided by running add_precursor_mz. For estimate_from_adduct=True this function will estimate the parent mass based on the mass and charge of known adducts. The table of known adduct properties can be found under
matchms/data/known_adducts_table.csv.- Parameters:
spectrum_in – Input spectrum.
estimate_from_adduct – When set to True, use adduct to estimate actual molecular mass (“parent mass”). Default is True. Switches back to charge-based estimate if adduct does not match a known adduct.
overwrite_existing_entry – Default is False. If set to True, a newly computed value will replace existing ones.
estimate_from_charge – Default is True. If set to True, the charge will be used to estimate the parent mass. Adduct of the form [M+H]+, [M+H]2+, [M-H]- etc are assumed.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added parent mass, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.add_precursor_formula(spectrum_in, clone: bool | None = True)[source]
Derive and set ‘precursor_formula’ from neutral ‘formula’ and ‘adduct’.
- Requirements:
spectrum_in must have metadata keys: ‘formula’ (neutral) and ‘adduct’.
‘formula’ must be a simple concatenation of element symbols and counts (no parentheses/hydrates/isotopes).
- matchms.filtering.add_precursor_mz(spectrum_in, clone: bool | None = True) Spectrum | None[source]
Add precursor_mz to correct field and make it a float.
For missing precursor_mz field: check if there is “pepmass”” entry instead. For string parsed as precursor_mz: convert to float.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added precursor mz metadata, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.add_retention_index(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Add retention index into ‘retention_index’ key if present.
- Parameters:
spectrum_in – Spectrum with RI information.
clone – Optionally clone the Spectrum.
- Return type:
Spectrum with RI info stored under ‘retention_index’.
- matchms.filtering.add_retention_time(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Add retention time information to the ‘retention_time’ key as float. Negative values and those not convertible to a float result in ‘retention_time’ being ‘None’.
- Parameters:
spectrum_in – Spectrum with retention time information.
clone – Optionally clone the Spectrum.
- Return type:
Spectrum with harmonized retention time information.
- matchms.filtering.clean_adduct(spectrum_in, clone: bool | None = True) Spectrum | None[source]
Clean adduct and make it consistent in style. Will transform adduct strings of type ‘M+H+’ to ‘[M+H]+’.
- Parameters:
spectrum_in – Matchms Spectrum object.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with cleaned adduct, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.clean_compound_name(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Clean compound name.
A list of frequently seen name additions that do not belong to the compound name will be removed.
- Parameters:
spectrum_in – Matchms Spectrum object.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with cleaned compound name, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.correct_charge(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Correct charge values based on given ionmode.
For some spectra, the charge value is either undefined or inconsistent with its ionmode, which is corrected by this filter.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with corrected charge derived from ionmode, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.default_filters(spectrum: Spectrum) Spectrum[source]
Collection of filters that are considered default and that do no require any (factory) arguments.
Collection is
- matchms.filtering.derive_adduct_from_name(spectrum_in: Spectrum, remove_adduct_from_name: bool = True, clone: bool | None = True) Spectrum | None[source]
Find adduct in compound name and add to metadata (if not present yet).
Method to interpret the given compound name to find the adduct.
- Parameters:
spectrum_in – Input spectrum.
remove_adduct_from_name – Remove found adducts from compound name if set to True. Default is True.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added adduct, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.derive_annotation_from_compound_name(spectrum_in: <module 'matchms.Spectrum' from '/home/docs/checkouts/readthedocs.org/user_builds/matchms/checkouts/latest/matchms/Spectrum.py'>, annotated_compound_names_file: str | None = None, mass_tolerance: float = 0.1, clone: bool | None = True) Spectrum | None[source]
Adds inchi, inchikey based on compound name by searching pubchem smiles is not supported anymore by pubchempy, see https://github.com/matchms/matchms/issues/823 Smiles can be derived from inchi, by running the filter derive_smiles_from_inchi
This filter is only run, if there is not yet a valid smiles or inchi in the metadata. The inchi and inchikey are only added if the found annotation is close enough to the parent mass.
- Parameters:
spectrum_in – The input spectrum.
annotated_compound_names_file (Optional[str]) – Any compound name that was searched for on pubchem will be added to this file. If a compound name is already in this file it will be used instead of looking up at pubchem. This file can be reused for future runs, speeding up the process. If None. The compound names found will still be cached for this run, but won’t be reusable for future runs. The csv file should contain the columns [“compound_name”, “smiles”, “inchi”, “inchikey”, “monoisotopic_mass”]
mass_tolerance – Acceptable mass difference between query compound and pubchem result.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added annotation, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.derive_formula_from_name(spectrum_in: Spectrum, remove_formula_from_name: bool = True, clone: bool | None = True) Spectrum | None[source]
Detect and remove misplaced formula in compound name and add to metadata.
Method to find misplaced formulas in compound name based on regular expression. This will not chemically test the detected formula, so the search is limited to frequently occuring types of shape ‘C47H83N1O8P1’.
- Parameters:
spectrum_in – Input spectrum.
remove_formula_from_name – Remove found formula from compound name if set to True. Default is True.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with cleaned formula, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.derive_formula_from_smiles(spectrum_in, overwrite=True, clone: bool | None = True) Spectrum | None[source]
Adds the molecule’s formula from SMILES.
- Parameters:
spectrum_in – Input spectrum.
overwrite – If True, will overwrite the formula. Default is True.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added molecular formular, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.derive_inchi_from_smiles(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Find missing Inchi and derive from smiles where possible.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added INCHI, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.derive_inchikey_from_inchi(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Find missing InchiKey and derive from Inchi where possible.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added INCHIKEY, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.derive_ionmode(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Derive missing ionmode based on adduct.
Some input formates (e.g. MGF files) do not always provide a correct ionmode. This function reads the adduct from the metadata and uses this to fill in the correct ionmode where missing.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Return type:
Spectrum object with ionmode attribute set.
- matchms.filtering.derive_smiles_from_inchi(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Find missing smiles and derive from Inchi where possible.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.#
- Returns:
Spectrum with added SMILES, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.harmonize_undefined_inchi(spectrum_in: Spectrum, undefined: str = '', aliases: List[str] = None, clone: bool | None = True) Spectrum | None[source]
Replace all aliases for empty/undefined inchi entries by value of
undefinedargument.- Parameters:
spectrum_in – Input spectrum.
undefined – Give desired entry for undefined inchi fields. Default is “”.
aliases – Enter list of strings that are expected to represent undefined entries. Default is [“”, “N/A”, “NA”, “n/a”].
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with undefined INCHI if not present or N/A, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.harmonize_undefined_inchikey(spectrum_in: Spectrum, undefined: str = '', aliases: List[str] = None, clone: bool | None = True) Spectrum | None[source]
Replace all aliases for empty/undefined inchikey entries by
undefined.- Parameters:
spectrum_in – Input spectrum.
undefined – Give desired entry for undefined inchikey fields. Default is “”.
aliases – Enter list of strings that are expected to represent undefined entries. Default is [“”, “N/A”, “NA”, “n/a”, “no data”].
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with undefined INCHIKEY if not present or N/A, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.harmonize_undefined_smiles(spectrum_in: Spectrum, undefined: str = '', aliases: List[str] = None, clone: bool | None = True) Spectrum | None[source]
Replace all aliases for empty/undefined smiles entries by
undefined.- Parameters:
spectrum_in – Input spectrum.
undefined – Give desired entry for undefined smiles fields. Default is “”.
aliases – Enter list of strings that are expected to represent undefined entries. Default is [“”, “N/A”, “NA”, “n/a”, “no data”].
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with undefined SMILES if not present or N/A, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.interpret_pepmass(spectrum_in, clone: bool | None = True) Spectrum | None[source]
Reads pepmass field (if present) and adds values to correct field(s).
The field “pepmass” or “PEPMASS” is often used to describe the precursor ion. This function will interpret the values as (mz, intensity, charge) tuple. Those will be splitted (if present) added to the fields “precursor_mz”, “precursor_intensity”, and “charge”.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with added pepmass, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.make_charge_int(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Convert charge field to integer (if possible).
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with converted charge, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.normalize_intensities(spectrum_in: Spectrum, clone: bool | None = True, scaling: tuple[float, float] | None = None) Spectrum | None[source]
Normalize intensities of peaks to unit height.
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
scaling – Optional tuple (min, max) to scale intensities to specific range. If None, normalizes to 0-1 range.
- Returns:
Spectrum with mormalized Intensities, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.reduce_to_number_of_peaks(spectrum_in: Spectrum, n_required: int = 0, n_max: int = inf, ratio_desired: float | None = None, clone: bool | None = True) Spectrum | None[source]
Lowest intensity peaks will be removed when it has more peaks than desired.
- Parameters:
spectrum_in – Input spectrum.
n_required – Number of minimum required peaks. Spectra with fewer peaks will be set to ‘None’. Default is 1.
n_max – Maximum number of peaks. Remove peaks if more peaks are found. Default is inf.
ratio_desired – Set desired ratio between maximum number of peaks and parent mass. For spectra without parent mass (e.g. GCMS spectra) this will raise an error when ratio_desired is used. Default is None.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with reduced lowest peaks, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.remove_noise_below_frequent_intensities(spectrum_in: Spectrum, min_count_of_frequent_intensities: int = 5, noise_level_multiplier: float = 2.0, clone: bool | None = True) Spectrum | None[source]
Removes noise if intensities exactly match frequently
When no noise filtering has been applied to a spectrum, many spectra show repeating intensities. From all intensities that repeat more than min_count_of_frequent_intensities the highest is selected. The noise level is set to this intensity * noise_level_multiplier. All fragments with an intensity below the noise level are removed.
This filter was suggested by Tytus Mak.
- Parameters:
spectrum_in – Input spectrum.
min_count_of_frequent_intensities – Minimum number of repeating intensities.
noise_level_multiplier – From all intensities that repeat more than min_count_of_frequent_intensities the highest is selected. The noise level is set to this intensity * noise_level_multiplier.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with removed intensities, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.remove_peaks_around_precursor_mz(spectrum_in: Spectrum, mz_tolerance: float = 17, clone: bool | None = True) Spectrum | None[source]
- Remove peaks that are within mz_tolerance (in Da) of
the precursor mz, excluding the precursor peak.
- Parameters:
spectrum_in – Input spectrum.
mz_tolerance – Tolerance of mz values that are not allowed to lie within the precursor mz. Default is 17 Da.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with removed peaks, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.remove_peaks_outside_top_k(spectrum_in: Spectrum, k: int = 6, mz_window: float = 50, clone: bool | None = True) Spectrum | None[source]
- Remove all peaks which are not within mz_window of at least one
of the k highest intensity peaks of the spectrum.
- Parameters:
spectrum_in – Input spectrum.
k – The number of most intense peaks to compare to. Default is 6.
mz_window – Window of mz values (in Da) that are allowed to lie within the top k peaks. Default is 50 Da.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with removed peaks, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.remove_peaks_relative_to_precursor_mz(spectrum_in: Spectrum, offset_to_precursor: float = -1.6, clone: bool | None = True) Spectrum | None[source]
Remove all peaks with m/z values > precursor-m/z + offset_to_precursor.
If offset_to_precursor is negative, this means that all peaks with m/z values greater than (precursor_mz - |offset_to_precursor|). If offset_to_precursor is positive, the precursor_mz peak itself will remain.
- Parameters:
spectrum_in – Input spectrum.
offset_to_precursor – All peaks with mz values > precursor_mz + offset_to_precursor will be removed. Default is -1.6 Da based Flash Entropy article by Li and Fiehn, 2023, Nat. Comm. (see https://www.nature.com/articles/s41592-023-02012-9)
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with removed peaks, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.remove_profiled_spectra(spectrum_in: Spectrum, mz_window=0.5, clone: bool | None = True) Spectrum | None[source]
Remove profiled spectra
Spectra are removed if within the mz_window of 0.5 of the highest peak at least 2 peaks next to the main peak are of intensity > max_intensity/2.
Reproduced from MZmine. https://github.com/mzmine/mzmine3/blob/master/src/main/java/io/github/mzmine/util/scans/ScanUtils.java#L609
- Parameters:
spectrum_in – Input spectrum.
mz_window – Window of mz values (in Da) that are allowed to lie within the top k peaks. Default is 50 Da.
clone – Optionally clone the Spectrum.
- Returns:
None if the spectrum is likely profile data, else the input spectrum.
- Return type:
Spectrum or None
- matchms.filtering.repair_adduct_and_parent_mass_based_on_smiles(spectrum_in: <module 'matchms.Spectrum' from '/home/docs/checkouts/readthedocs.org/user_builds/matchms/checkouts/latest/matchms/Spectrum.py'>, mass_tolerance: float, clone: bool | None = True) Spectrum | None[source]
Corrects the adduct and parent mass of a spectrum based on its SMILES representation and the precursor m/z.
Given a spectrum, this function tries to match the spectrum’s parent mass, derived from its precursor m/z and known adducts, to the neutral monoisotopic mass of the molecule derived from its SMILES representation. If a match is found within a given mass tolerance, the adduct and parent mass of the spectrum are updated.
Parameters:
- spectrum_inSpectrum
The input spectrum whose adduct needs to be repaired.
- mass_tolerancefloat
Maximum allowed mass difference between the calculated parent mass and the neutral monoisotopic mass derived from the SMILES.
- clone:
Optionally clone the Spectrum.
- returns:
Spectrum with repaired parent mass, or None if not present.
- rtype:
Spectrum or None
- matchms.filtering.repair_adduct_based_on_parent_mass(spectrum_in: <module 'matchms.Spectrum' from '/home/docs/checkouts/readthedocs.org/user_builds/matchms/checkouts/latest/matchms/Spectrum.py'>, mass_tolerance: float, clone: bool | None = True) Spectrum | None[source]
Corrects the adduct of a spectrum based on its parent_mass representation and the precursor m/z.
Parameters:
- spectrum_inSpectrum
The input spectrum whose adduct needs to be repaired.
- mass_tolerancefloat
Maximum allowed mass difference between the parent mass and the parent mass based on the adduct.
- clone:
Optionally clone the Spectrum.
- returns:
Spectrum with repaired parent adduct, or None if not present.
- rtype:
Spectrum or None
- matchms.filtering.repair_inchi_inchikey_smiles(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
Check if inchi, inchikey, and smiles entries seem correct. Detect and correct if any of those entries clearly belongs into one of the other two fields (e.g. inchikey found in inchi field).
- Parameters:
spectrum_in – Input spectrum.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with repaired INCHI, INCHIKEY and SMILES, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.repair_not_matching_annotation(spectrum_in: <module 'matchms.Spectrum' from '/home/docs/checkouts/readthedocs.org/user_builds/matchms/checkouts/latest/matchms/Spectrum.py'>, clone: bool | None = True) Spectrum | None[source]
Repairs mismatches in a spectrum’s annotations related to SMILES, InChI, and InChIKey.
Given a spectrum, this function ensures that the provided SMILES, InChI, and InChIKey annotations are consistent with one another. If there are discrepancies, they are resolved as follows:
- If the SMILES and InChI do not match:
Both SMILES and InChI are checked against the parent mass.
The annotation that matches the parent mass is retained, and the other is regenerated.
- If the InChIKey does not match the InChI:
A new InChIKey is generated from the InChI and replaces the old one.
Warnings and information logs are generated to track changes and potential issues. For correctness of InChIKey entries, only the first 14 characters are considered.
Parameters:
- spectrum_inSpectrum
The input spectrum containing annotations to be checked and repaired.
- clone:
Optionally clone the Spectrum.
Returns:
- Spectrum
A cloned version of the input spectrum with corrected annotations. If the input spectrum is None, it returns None.
- matchms.filtering.repair_parent_mass_from_smiles(spectrum_in: <module 'matchms.Spectrum' from '/home/docs/checkouts/readthedocs.org/user_builds/matchms/checkouts/latest/matchms/Spectrum.py'>, mass_tolerance: float = 0.1, clone: bool | None = True) Spectrum | None[source]
Sets the parent mass to match the smiles mass, if not already close to smiles mass
Parameters:
- spectrum_inSpectrum
The input spectrum containing annotations to be checked and repaired.
- clone:
Optionally clone the Spectrum.
- returns:
Spectrum with repaired parent mass, or None if not present.
- rtype:
Spectrum or None
- matchms.filtering.repair_parent_mass_is_molar_mass(spectrum_in: <module 'matchms.Spectrum' from '/home/docs/checkouts/readthedocs.org/user_builds/matchms/checkouts/latest/matchms/Spectrum.py'>, mass_tolerance: float, clone: bool | None = True) Spectrum | None[source]
Changes the parent mass from molar mass into monoistopic mass
Manual entered parent mass is sometimes wrongly added as Molar mass instead of monoisotopic mass We check if the given parent mass is equal to the Molar mass (based on the smiles) and correct it to the monoisotopic mass in these cases.
The molar mass is an average mass based on the average of all common isotopes and will therefore differ from what is measured in mass spectrometry.
Parameters:
- spectrum_inSpectrum
The input spectrum containing annotations to be checked and repaired.
- mass_tolerance:
Maximum allowed mass difference between the calculated parent mass and the neutral monoisotopic mass derived from the SMILES.
- clone:
Optionally clone the Spectrum.
- returns:
Spectrum with repaired parent mass, or None if not present.
- rtype:
Spectrum or None
- matchms.filtering.repair_parent_mass_match_smiles_wrapper(spectrum_in: Spectrum, mass_tolerance: float = 0.2, clone: bool | None = True) Spectrum | None[source]
Wrapper function for repairing a mismatch between parent mass and smiles mass
Parameters:
- spectrum_inSpectrum
The input spectrum containing annotations to be checked and repaired.
- mass_tolerance:
Maximum allowed mass difference between the calculated parent mass and the neutral monoisotopic mass derived from the SMILES. Defaults to 0.2.
- clone:
Optionally clone the Spectrum.
- returns:
Spectrum with repaired parent mass, or None if not present.
- rtype:
Spectrum or None
- matchms.filtering.repair_smiles_of_salts(spectrum_in, mass_tolerance, clone: bool | None = True) Spectrum | None[source]
Repairs the smiles of a salt to match the parent mass. E.g. C1=NC2=NC=NC(=C2N1)N.Cl is converted to 1=NC2=NC=NC(=C2N1)N if this matches the parent mass Checks if parent mass matches one of the ions
- Parameters:
spectrum_in – Input spectrum.
mass_tolerance – Maximum allowed mass difference between the calculated parent mass and the neutral monoisotopic mass derived from the SMILES.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with repaired SMILES, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.require_compound_name(spectrum: Spectrum) Spectrum | None[source]
Ensure that the compound name is present in the spectrum metadata.
- matchms.filtering.require_correct_ionmode(spectrum_in: Spectrum, ion_mode_to_keep) Spectrum | None[source]
Validates the ion mode of a given spectrum. If the spectrum’s ion mode doesn’t match the ion_mode_to_keep, it will be removed and a log message will be generated.
- Parameters:
- Returns:
The validated spectrum if its ion mode matches the desired one, or None otherwise.
- Return type:
Spectrum or None
- matchms.filtering.require_formula(spectrum: Spectrum) Spectrum | None[source]
Ensure that the molecular formula is present and looks like a valid formula.
- matchms.filtering.require_matching_adduct_precursor_mz_parent_mass(spectrum, tolerance=0.1) Spectrum | None[source]
Checks if the adduct precursor mz and parent mass match within the tolerance
- matchms.filtering.require_maximum_number_of_peaks(spectrum_in: Spectrum, maximum_number_of_fragments: int = 1000, clone: bool | None = True) Spectrum | None[source]
Spectrum will be set to None when it has more peaks than maximum_number_of_fragments.
- Parameters:
spectrum_in – Input spectrum.
maximum_number_of_fragments – Number of minimum required peaks. Spectra with fewer peaks will be set to ‘None’.
clone – Optionally clone the Spectrum.
- Returns:
Untouched Spectrum or ‘None’.
- Return type:
Spectrum or None
- matchms.filtering.require_minimum_number_of_high_peaks(spectrum_in: Spectrum, no_peaks: int = 5, intensity_percent: float = 2.0, clone: bool | None = True) Spectrum | None[source]
- Returns None if the number of peaks with relative intensity
above or equal to intensity_percent is less than no_peaks.
- Parameters:
spectrum_in – Input spectrum.
no_peaks – Minimum number of peaks allowed to have relative intensity above intensity_percent. Less peaks will return none. Default is 5.
intensity_percent – Minimum relative intensity (as a percentage between 0-100) for peaks that are searched. Default is 2.
clone – Optionally clone the Spectrum.
- Returns:
Untouched Spectrum or ‘None’.
- Return type:
Spectrum or None
- matchms.filtering.require_minimum_number_of_peaks(spectrum_in: Spectrum, n_required: int = 10, ratio_required: float | None = None, clone: bool | None = True) Spectrum | None[source]
Spectrum will be set to None when it has fewer peaks than required.
- Parameters:
spectrum_in – Input spectrum.
n_required – Number of minimum required peaks. Spectra with fewer peaks will be set to ‘None’.
ratio_required – Set desired ratio between minimum number of peaks and parent mass. Default is None.
clone – Optionally clone the Spectrum.
- Returns:
Untouched Spectrum or ‘None’.
- Return type:
Spectrum or None
- matchms.filtering.require_parent_mass_match_smiles(spectrum_in: Spectrum, mass_tolerance) Spectrum | None[source]
Validates if the parent mass of the given spectrum matches the mass calculated from its associated SMILES string within a specified tolerance.
- Parameters:
- Returns:
The validated spectrum if its parent mass matches the SMILES mass within the specified tolerance, or None otherwise.
- Return type:
Spectrum or None
- matchms.filtering.require_precursor_below_mz(spectrum_in: Spectrum, max_mz: float = 1000) Spectrum[source]
- Returns None if the precursor_mz of a spectrum is above
max_mz.
- Parameters:
spectrum_in – Input spectrum.
max_mz – Maximum mz value for the precursor mz of a spectrum. All precursor mz values greater or equal to this will return none. Default is 1000.
- matchms.filtering.require_precursor_mz(spectrum_in: Spectrum, minimum_accepted_mz: float | None = 10.0, maximum_mz: float | None = None, clone: bool | None = True) Spectrum | None[source]
Returns None if there is no precursor_mz or if <= minimum_accepted_mz
- Parameters:
spectrum_in – Input spectrum.
minimum_accepted_mz – Set to minimum acceptable value for precursor m/z. Default is set to 10.0.
maximum_mz – Set the maximum value for precursor m/z.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with precursor_mz, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.require_retention_index(spectrum_in: Spectrum, clone: bool | None = True) Spectrum | None[source]
This function checks if the input spectrum has a ‘retention_index’ in its metadata. If the input spectrum is None or doesn’t have a ‘retention_index’, the function returns None. Otherwise, it returns a clone of the input spectrum.
- Parameters:
(SpectrumType) (spectrum_in) – The input spectrum to check.
clone – Optionally clone the Spectrum.
Returns
SpectrumType (A clone of the input spectrum if it has a 'retention_index', None otherwise.)
- matchms.filtering.require_retention_time(spectrum_in: Spectrum, minimum_rt=None, maximum_rt=None, clone: bool | None = True) Spectrum | None[source]
This function checks if the input spectrum has a ‘retention_time’ in its metadata. If the input spectrum is None or doesn’t have a ‘retention_time’, the function returns None. Otherwise, it returns a clone of the input spectrum.
- Parameters:
(SpectrumType) (spectrum_in) – The input spectrum to check.
clone – Optionally clone the Spectrum.
Returns
SpectrumType (A clone of the input spectrum if it has a 'retention_time', None otherwise.)
- matchms.filtering.require_valid_annotation(spectrum: <module 'matchms.Spectrum' from '/home/docs/checkouts/readthedocs.org/user_builds/matchms/checkouts/latest/matchms/Spectrum.py'>) Spectrum | None[source]
Removes spectra that are not fully annotated (correct and matching, smiles, inchi and inchikey)
- matchms.filtering.select_by_intensity(spectrum_in: Spectrum, intensity_from: float = 10.0, intensity_to: float = 200.0, clone: bool | None = True) Spectrum | None[source]
Keep only peaks within set intensity range (keep if intensity_from >= intensity >= intensity_to). In most cases it is adviced to use
select_by_relative_intensity()function instead.- Parameters:
spectrum_in – Input spectrum.
intensity_from – Set lower threshold for peak intensity. Default is 10.0.
intensity_to – Set upper threshold for peak intensity. Default is 200.0.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with peaks within the specified intensity range, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.select_by_mz(spectrum_in: Spectrum, mz_from: float = 0.0, mz_to: float = 1000.0, clone: bool | None = True) Spectrum | None[source]
Keep only peaks between mz_from and mz_to (keep if mz_from >= m/z >= mz_to).
- Parameters:
spectrum_in – Input spectrum.
mz_from – Set lower threshold for m/z peak positions. Default is 0.0.
mz_to – Set upper threshold for m/z peak positions. Default is 1000.0.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with peaks within the specified mz range, or None if not present.
- Return type:
Spectrum or None
- matchms.filtering.select_by_relative_intensity(spectrum_in: Spectrum, intensity_from: float = 0.0, intensity_to: float = 1.0, clone: bool | None = True) Spectrum | None[source]
Keep only peaks within set relative intensity range (keep if intensity_from >= intensity >= intensity_to).
- Parameters:
spectrum_in – Input spectrum.
intensity_from – Set lower threshold for relative peak intensity. Default is 0.0.
intensity_to – Set upper threshold for relative peak intensity. Default is 1.0.
clone – Optionally clone the Spectrum.
- Returns:
Spectrum with peaks within the relative intensity range, or None if not present.
- Return type:
Spectrum or None
Subpackages
- matchms.filtering.filter_utils package
- Submodules
- matchms.filtering.filter_utils.derive_precursor_mz_and_parent_mass module
- matchms.filtering.filter_utils.get_neutral_mass_from_smiles module
- matchms.filtering.filter_utils.interpret_unknown_adduct module
- matchms.filtering.filter_utils.load_known_adducts module
- matchms.filtering.filter_utils.smile_inchi_inchikey_conversions module
- Submodules
- matchms.filtering.metadata_processing package
- Submodules
- matchms.filtering.metadata_processing.add_compound_name module
- matchms.filtering.metadata_processing.add_fingerprint module
- matchms.filtering.metadata_processing.add_parent_mass module
- matchms.filtering.metadata_processing.add_precursor_formula module
- matchms.filtering.metadata_processing.add_precursor_mz module
- matchms.filtering.metadata_processing.add_retention module
- matchms.filtering.metadata_processing.clean_adduct module
- matchms.filtering.metadata_processing.clean_compound_name module
- matchms.filtering.metadata_processing.correct_charge module
- matchms.filtering.metadata_processing.derive_adduct_from_name module
- matchms.filtering.metadata_processing.derive_annotation_from_compound_name module
- matchms.filtering.metadata_processing.derive_formula_from_name module
- matchms.filtering.metadata_processing.derive_formula_from_smiles module
- matchms.filtering.metadata_processing.derive_inchi_from_smiles module
- matchms.filtering.metadata_processing.derive_inchikey_from_inchi module
- matchms.filtering.metadata_processing.derive_ionmode module
- matchms.filtering.metadata_processing.derive_smiles_from_inchi module
- matchms.filtering.metadata_processing.harmonize_undefined_inchi module
- matchms.filtering.metadata_processing.harmonize_undefined_inchikey module
- matchms.filtering.metadata_processing.harmonize_undefined_smiles module
- matchms.filtering.metadata_processing.interpret_pepmass module
- matchms.filtering.metadata_processing.make_charge_int module
- matchms.filtering.metadata_processing.repair_adduct_and_parent_mass_based_on_smiles module
- matchms.filtering.metadata_processing.repair_adduct_based_on_parent_mass module
- matchms.filtering.metadata_processing.repair_inchi_inchikey_smiles module
- matchms.filtering.metadata_processing.repair_not_matching_annotation module
- matchms.filtering.metadata_processing.repair_parent_mass_from_smiles module
- matchms.filtering.metadata_processing.repair_parent_mass_is_molar_mass module
- matchms.filtering.metadata_processing.repair_parent_mass_match_smiles_wrapper module
- matchms.filtering.metadata_processing.repair_smiles_of_salts module
- matchms.filtering.metadata_processing.require_compound_name module
- matchms.filtering.metadata_processing.require_correct_ionmode module
- matchms.filtering.metadata_processing.require_correct_ms_level module
- matchms.filtering.metadata_processing.require_formula module
- matchms.filtering.metadata_processing.require_matching_adduct_and_ionmode module
- matchms.filtering.metadata_processing.require_matching_adduct_precursor_mz_parent_mass module
- matchms.filtering.metadata_processing.require_parent_mass_match_smiles module
- matchms.filtering.metadata_processing.require_precursor_mz module
- matchms.filtering.metadata_processing.require_retention_index module
- matchms.filtering.metadata_processing.require_retention_time module
- matchms.filtering.metadata_processing.require_valid_annotation module
- Submodules
- matchms.filtering.peak_processing package
- Submodules
- matchms.filtering.peak_processing.normalize_intensities module
- matchms.filtering.peak_processing.reduce_to_number_of_peaks module
- matchms.filtering.peak_processing.remove_noise_below_frequent_intensities module
- matchms.filtering.peak_processing.remove_peaks_around_precursor_mz module
- matchms.filtering.peak_processing.remove_peaks_outside_top_k module
- matchms.filtering.peak_processing.remove_peaks_relative_to_precursor_mz module
- matchms.filtering.peak_processing.remove_profiled_spectra module
- matchms.filtering.peak_processing.require_maximum_number_of_peaks module
- matchms.filtering.peak_processing.require_minimum_number_of_high_peaks module
- matchms.filtering.peak_processing.require_minimum_number_of_peaks module
- matchms.filtering.peak_processing.select_by_intensity module
- matchms.filtering.peak_processing.select_by_mz module
- matchms.filtering.peak_processing.select_by_relative_intensity module
- Submodules
Submodules
- matchms.filtering.SpeciesString module
SpeciesStringSpeciesString.dirtySpeciesString.targetSpeciesString.cleanedSpeciesString.__init__()SpeciesString.clean()SpeciesString.clean_as_inchi()SpeciesString.clean_as_inchikey()SpeciesString.clean_as_smiles()SpeciesString.guess_target()SpeciesString.looks_like_a_smiles()SpeciesString.looks_like_an_inchi()SpeciesString.looks_like_an_inchikey()
- matchms.filtering.SpectrumProcessor module
- matchms.filtering.default_filters module
- matchms.filtering.default_pipelines module
- matchms.filtering.filter_order module